Background: Hierarchy of interactions leads to aelf-organizinglevels of structure
The complex 3-dimensional structure of biological macromolecules reflects a hierarchy of underlying physical interactions. Covalent bonding guarantees the stability and local geometry of the primary structure, i.e. the sequence of aminoacids in proteins or bases in DNA. Weaker (hydrogen bonding, hydrophobic and base stacking) interactions, whose energies are roughly comparable with the thermal content of the surroundings, are ultimately responsible for the formation of higher-level structures associated with biological function.
Mesoscopic modeling adopts a minimalist approach. It foregoes any attempt to describe the details of structure and/or dynamics and concentrates instead on those gross features of a biological macromolecule which characterize collective behavior. Such cooperativity manifests itself in macroscopic phenomena, e.g. protein (or DNA) denaturation.
An example of this minimalist approach is the nonlinear lattice dynamics model of DNA proposed by Peyrard, Bishop and Dauxois (PBD). In its simplest version the model looks at a base pair as a single, continuous degree of freedom. Thermodynamic behavior is governed by the interplay between binding forces. The model describes successfully the phenomena of thermal and mechanical denaturation of long DNA chains (commonly known as melting and unzipping, respectively).
Single strands of DNA with complementary ends will spontaneously self-assemble to form "hairpin" structures. Hairpins provide a unique experimental (and theoretical) laboratory to test DNA collective dynamics.
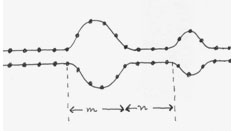
The double helix is in reality an open, fluctuating structure. Local openings of base pairs are instrumental in initiating the process of transcription. Understanding DNA bubble statistics and dynamics is an important task of biological physics.
A detailed analysis along the lines of the PBD model reveals that it takes a large amount of energy to open the first few sites of a bubble; once the "initiation barrier" has been crossed, bubble growth proceeds with a very low energetic cost.
Melting profiles
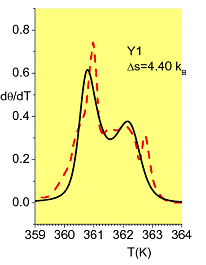
With an improved set of global parameters and a novel, efficient computational procedure, it is possible to use the PBD model in order to predict the melting profile of any given long, genomic sequence.
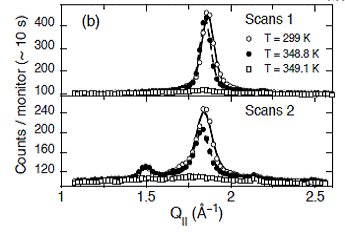
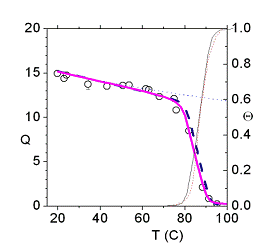
The thermal denaturation of DNA studied with neutron scattering, A. Wildes, N. Theodorakopoulos, J. Valle-Orero, S. Cuesta-Lopez, J-L Garden and M. Peyrard, Phys. Rev. Lett. 106, 048101 (2011);arXiv:1101.1797
DNA "breathing" flexibility
Understanding the temperature (and sequence) dependence of DNA bending stiffness - an essential property in key biological processes - is now possible in terms of a combined PBD and Kratky-Porod (KP) model.
Contact N. Theodorakopoulos
Theoretical and Physical Chemistry Institute,
National Hellenic Research Foundation,
48 Vassileos Constantinou Ave.,
Athens 11635, Greece